4 SNV calling
Bisulfite sequencing data contains information of both methylation and genome sequences. In addition to DNA methylation analysis, we can also call variants using bisulfite data. Due to bisulfite coversion and PCR amplification during library preparation, the unmethylated cytosines on the DNA fragments would be converted to thymines. Thus, it’s difficult to distinguish thymine produced by bisulfite coversion with the real thymine allele.
In recent years, few tools are adapted to bisulfite data for SNP calling. The main idea is removing vague reads that may contain unmethylated cytosines for a given positoin. Consequently, the rest reads can be regarded as reads generated from a normal genome DNA without bisulfite treatment and can be used to call variants using regular methods without consideration of bisulfite conversion.
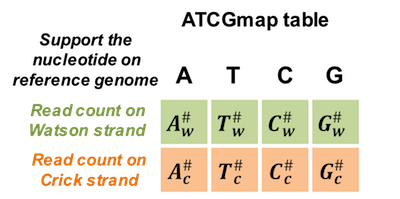
Figure 4.1: ATCGmap table used for SNV calling
However, removing the vague reads leads to information lost in most cases making variant calling less confident, especially when the sequencing depth is low.
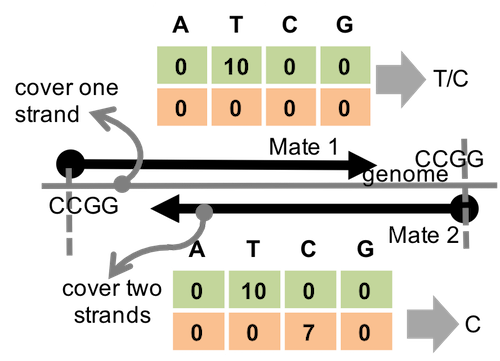
Figure 4.2: Example that alignments refer to vague genotypes
To solve this problem, we tried to introduce wild-card in genotype calling. Even for these amiguouse genotypes, we can still learning something.
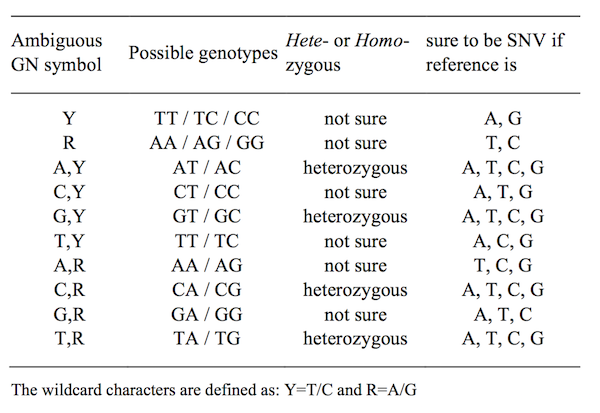
Figure 4.3: Table for definition of amibiguous genotype
We proposed two independent methods called BinomWC (based on binomial) and BayesWC (based on bayesian), taking vague reads into consideration.
4.1 BaysWC strategy
BinomWC strategy combines bayesian method and wildcard strategy for predicting the genotype. The likelyhood matrix is designed as following.

Figure 4.4: The likelyhood matrix for BayesWC strategy
4.2 BinomWC strategy
BinomWC (Binomial-WildCard) strategy works as following for 3 cases.
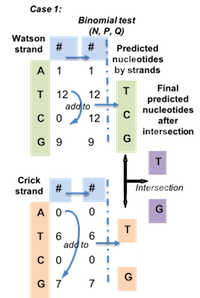
Figure 4.5: Case 1 for BinomWC strategy
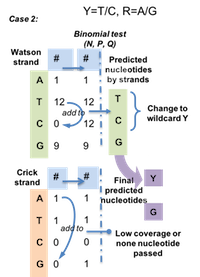
Figure 4.6: Case 2 for BinomWC strategy
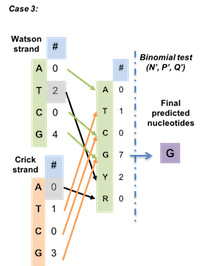
Figure 4.7: Case 3 for BinomWC strategy
4.3 Performance
Performances on simulation data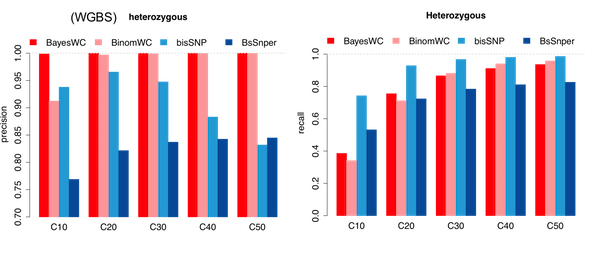
Figure 4.8: Precision-Recall analysis on simulated WGBS data
Command
cgmaptools snv -h# Usage: cgmaptools snv [-i <ATCGmap>] [-o <output> -v <VCF>]
# (aka SNVFromATCGmap)
# Description: Predict the SNV from ATCGmap file.
# Contact: Guo, Weilong; guoweilong@126.com
# Last update: 2018-05-02
# Output format example:
# #chr nuc pos ATCG_watson ATCG_crick predicted_nuc p_value
# chr1 G 4752 17,0,0,69 0,0,0,0 A,G 9.3e-07
# chr1 A 4770 40,0,0,29 0,0,0,0 A,G 0.0e+00
# chr1 T 8454 0,39,0,0 0,0,0,0 T/C 1.00e-01
#
#
# Options:
# -h, --help show this help message and exit
# -i FILE ATCGmap format, STDIN if not specified
# -v FILE, --vcf=FILE VCF format file for output
# -a, --all_nt Show all sites with an predictable genotype. Only show
# SNP sites if not specified.
# -o OUTFILE STDOUT if not specified
# -m MODE, --mode=MODE Mode for calling SNP [Default: binom]
# binom: binomial, separate strands
# bayes: bayesian mode
# --bayes-e=BAYES_ER (BayesWC mode) Error rate for calling a nucleotide
# [Default: 0.05]
# --bayes-p=BAYES_PV (BayesWC mode) P value as cut-off [Default: 0.001]
# --bayes-dynamicP (BayesWC mode) Use dynamic p-value for different
# coverages install of specific p-value. (Recomended)
# "--bayes-p" will be ignored if "--bayes-dynamicP" is
# specified.
# --binom-e=BINOM_ER (BinomWC mode) Error rate for calling a nucleotide
# [Default: 0.05]
# --binom-p=BINOM_PV (BinomWC mode) P value as cut-off [Default: 0.01]
# --binom-cov=BINOM_COV
# (BinomWC mode) The coverage checkpoint [Default: 10]Example commands :
cgmaptools snv -i WG.ATCGmap.gz -m bayes -v bayes.vcf -o bayes.snv --bayes-dynamicPcgmaptools snv -i WG.ATCGmap.gz -m binom -o binom.snvOutput format
- Example
#chr nuc pos ATCG_watson ATCG_crick predicted_nuc p_value chr1 G 4752 17, 0, 0, 69 0, 0, 0, 0 A,G 9.3e-07 chr1 A 4770 40, 0, 0, 29 0, 0, 0, 0 A,G 0.0e+00 chr1 T 8454 0, 39, 0, 0 0, 0, 0, 0 T/C 1.00e-01- Column Description

Figure 4.9: Output format description for cgmaptools snv