3 File Manipulation
CGmapTools provides multiple utilities to manipulate files in ATCGmap and CGmap format or compressed ATCGbz/CGbz format.
Usage: cgmaptools <convert|fetch|refill|intersect|merge2|mergelist|sort|split|select|> [options]
3.1 convert
Description : File format coversion.
Table of command for converting formats:
| Commands | From | To |
|---|---|---|
| bam2cgmap | BAM | CGmap & ATCGmap |
| atcgmap2atcgbz | ATCGmap | ATCGbz |
| atcgbz2atcgmap | ATCGbz | ATCGmap |
| atcgmap2cgmap | ATCGmap | CGmap |
| cgmap2cgbz | CGamp | CGbz |
| cgbz2cgmap | CGbz | CGmap |
| cgmap2wig | CGmap | WIG |
| bismark2cgmap | Bismark | CGmap |
- Command
cgmaptools convert -h # Usage: cgmaptools convert <command> [options]
# Version: 0.1.2
# Updated on: Dec. 14th, 2018
# Commands:
# bam2cgmap BAM => CGmap & ATCGmap
# atcgmap2atcgbz ATCGmap => ATCGbz
# atcgbz2atcgmap ATCGbz => ATCGmap
# atcgmap2cgmap ATCGmap => CGmap
# cgmap2cgbz CGamp => CGbz
# cgbz2cgmap CGbz => CGmap
# cgmap2wig CGmap => WIG
# bismark2cgmap Bismark => CGmapExample :
- BAM to CGmap
cgmaptools convert bam2cgmap -b WG.bam -g genome.fa --rmOverlap -o WG- BAM to CGmap
cgmaptools convert bam2cgmap -b RR.bam -g genome.fa --rmOverlap -o RR- ATCGmap to ATCGbz
cgmaptools convert atcgmap2atcgbz -c WG.ATCGmap.gz -b WG.ATCGbz- ATCGvz to ATCGmap
cgmaptools convert atcgbz2atcgmap -c WG2.ATCGmap.gz -b WG.ATCGbz- CGmap to CGbz
cgmaptools convert cgmap2cgbz -c RR.CGmap.gz -b RR.CGbz- CGbz to CGmap
cgmaptools convert cgbz2cgmap -c RR2.CGmap.gz -b RR.CGbz- CGmap to WIG
cgmaptools convert cgmap2wig -i <CGmap> [-w <wig>] [-c <INT> -b <float>]- bismark output to CGmap
cgmaptools convert bismark2cgmap -i bismark.dat -o output.CGmap
Note: please refer to the help message for usage details using
-hoption.
3.2 fetch
Description: Fastly acess methylation data in specified region.
Command
cgmaptools fetch -h# Usage: cgmaptools fetch <command> [options]
# Version: 0.1.2
# Updated on: Dec. 14th, 2018
# Commands:
# atcgbz fetch lines from ATCGbz
# cgbz fetch lines from CGbz3.2.1 fetch cgbz
- Command
cgmaptools fetch cgbz -h#
# Usage: cgmaptools fetch cgbz -b <CGbz> -C <CHR> -L <LeftPos> -R <RightPos>
# (aka CGvzFetchRegion)
# Description: Convert CGbz file to CGmap format.
# Contact: Guo, Weilong; guoweilong@126.com
# Last update: 2016-12-07
#
# Options:
#
# -h, --help output help information
# -b, --CGbz <arg> output CGbz file
# -C, --CHR <arg> specify the chromosome name
# -L, --leftPos <arg> the left position
# -R, --rightPos <arg> the right positionExample :
cgmaptools fetch cgbz -b RR.CGbz -C chr3 -L 2200 -R 2400
3.2.2 fetch atcgbz
- Command
cgmaptools fetch atcgbz -h#
# Usage: cgmaptools fetch atcgbz -b <ATCGbz> -C <CHR> -L <LeftPos> -R <RightPos>
# (aka ATCGbzFetchRegion)
# Description: Convert ATCGbz format to ATCGmap format.
# Contact: Guo, Weilong; guoweilong@126.com
# Last update: 2016-12-07
#
# Options:
#
# -h, --help output help information
# -b, --ATCGbz <arg> output ATCGbz file
# -C, --CHR <arg> specify the chromosome name
# -L, --leftPos <arg> the left position
# -R, --rightPos <arg> the right positionExample :
cgmaptools fetch atcgbz -b WG.ATCGbz -C chr2 -L 90 -R 100
3.3 refill
- Command
cgmaptools refill -h# Usage: cgmaptools refill [-i <CGmap>] -g <genome.fa> [-o output]
# (aka CGmapFillContext)
# Description: Fill the CG/CHG/CHH and CA/CC/CT/CG context.
# Other fields will not be affected.
# Can be applied to ATCGmap file.
# Contact: Guo, Weilong; guoweilong@126.com;
# Last Update: 2018-01-02
# Index Ex:
# Chr1 C 3541 - - 0.0 0 1
# Output Ex:
# Chr1 C 3541 CG CG 0.0 0 1
#
# Options:
# -h, --help show this help message and exit
# -i STRING Input CGmap file (CGmap or CGmap.gz)
# -g STRING genome file, FASTA format (gzipped if end with '.gz')
# -o STRING Output file name (gzipped if end with '.gz')
# -0, --0-base 0-based genome if specified [Default: 1-based]File formats:
The input CGmap file, which is lacking C context on the 3rd and 4th columns:
Chr1 C 3541 - - 0.0 0 1After
refillprocessing, the CGmap file would be as below, added C context information:Chr1 C 3541 CG CG 0.0 0 1Example:
zcat RR2.CGmap.gz | gawk -F"\t" -vOFS="\t" '{$4="-"; $5="-"; print;}' | cgmaptools refill -g genome.fa -o RR3.CGmap.gz
3.4 intersect
- Command
cgmaptools intersect -h# Usage: cgmaptools intersect [-1 <CGmap_1>] -2 <CGmap_2> [-o <output>]
# (aka CGmapIntersect)
# Description:
# Get the intersection of two CGmap files.Contact: Guo, Weilong; guoweilong@126.com
# Last Update: 2018-04-10
# Output Format:
# Chr1 C 3541 CG CG 0.8 4 5 0.4 4 10
# When 1st CGmap file is:
# Chr1 C 3541 CG CG 0.8 4 5
# ,and 2nd CGmap file is:
# Chr1 C 3541 CG CG 0.4 4 10
#
# Options:
# -h, --help show this help message and exit
# -1 CGmap File File name, end with .CGmap or .CGmap.gz.
# -2 CGmap File standard input if not specified
# -o OUTFILE To standard output if not specified. Compressed output
# if end with .gz
# -C CONTEXT, --context=CONTEXT
# specific context: CG, CH, CHG, CHH, CA, CC, CT, CW
# use all sites if not specifiedExample
cgmaptools intersect -1 WG.CGmap.gz -2 RR.CGmap.gz -C CG -o intersect_CG.gzOutput format
- Example
Chr1 C 3541 CG CG 0.8 4 5 0.4 4 10 Chr1 C 3542 CG CG 0.8 3 5 0.2 2 10 Chr1 C 3545 CHG CA 0.0 0 5 0.1 1 10- Column Description

Figure 3.1: Output format description for cgmaptools intersect
3.5 merge2
Command
cgmaptools merge2 -h# Usage: cgmaptools merge2 <command> [options]
# Version: 0.1.2
# Updated on: Dec. 14th, 2018
# Commands:
# atcgmap merge two ATCGmap files into one
# cgmap merge two CGmap files into one3.5.1 merge2 atcgmap
Command
cgmaptools merge2 atcgmap -h# Unknown option: -h
# Usage: cgmaptools merge2 atcgmap -1 <ATCGmap> -2 <ATCGmap>
# (aka ATCGmapMerge)
# Contact: Guo, Weilong; guoweilong@126.com;
# Last Update: 2016-12-07
# Options:
# -1 Input, 1st ATCGmap file
# -2 Input, 2nd ATCGmap file
# Output to STDOUT in ATCGmap format
# Tips: Two input files should have the same order of chromosomesExample
cgmaptools merge2 atcgmap -1 WG.ATCGmap.gz -2 RR.ATCGmap.gz | gzip > merge.ATCGmap.gz
3.5.2 merge2 cgmap
Command
cgmaptools merge2 cgmap -h# Usage: cgmaptools merge2 cgmap -1 <CGmap_1> -2 <CGmap_2> [-o <output>]
# (aka CGmapMerge)
# Description: Merge two CGmap files together.
# Contact: Guo, Weilong; guoweilong@126.com
# Last Update: 2018-01-02
# Note: The two input CGmap files should be sorted in the same order first.
#
#
# Options:
# -h, --help show this help message and exit
# -1 FILE File name end with .CGmap or .CGmap.gz
# -2 FILE If not specified, STDIN will be used.
# -o OUTFILE CGmap, output file. Use STDOUT if omitted (gzipped if end with
# '.gz').Example
Example command :
cgmaptools merge2 cgmap -1 WG.CGmap.gz -2 RR.CGmap.gz | gzip > merge.CGmap.gz
3.6 mergelist
- Command
cgmaptools mergelist -h# Usage: cgmaptools mergelist <command> [options]
# Version: 0.1.2
# Updated on: Dec. 14th, 2018
# Commands:
# tomatrix mC levels matrix from multiple files
# tosingle merge list of input files into one3.6.1 mergelist tomatrix
- Command
cgmaptools mergelist tomatrix -h# Usage: cgmaptools mergelist tomatrix [-i <index>] -f <IN1,IN2,..> -t <tag1,tag2,..> [-o output]
# (aka CGmapFillIndex)
# Description: Fill methylation levels according to the Index file for CGmap files in list.
# Contact: Guo, Weilong; guoweilong@126.com;
# Last Updated: 2018-05-02
# Index format Ex:
# chr10 100005504
# Output format Ex:
# chr pos tag1 tag2 tag3
# Chr1 111403 0.30 nan 0.80
# Chr1 111406 0.66 0.40 0.60
#
# Options:
# -h, --help show this help message and exit
# -i FILE TXT file, index file, use STDIN if omitted
# -f STRING List of (input) CGmap files (CGmap or CGmap.gz)
# -t STRING List of tags, same order with '-f'
# -c INT minimum coverage [default: 1]
# -C INT maximum coverage [default: 200]
# -o STRING Output file name (gzipped if end with '.gz')Example
zcat RR*.CGmap.gz WG.CGmap.gz | gawk '$8>=5' | cut -f1,3 | sort -u | cgmaptools sort -c 1 -p 2 > indexcgmaptools mergelist tomatrix -i index -f RR.CGmap.gz,RR2.CGmap.gz,WG.CGmap.gz -t RR,RR2,WG -c 5 -C 100 -o matrix.CG.gzFormat for Index file
- Example
Chr1 940 Chr1 1840 Chr2 9060- Column Description

Figure 3.2: Format description for INDEX file
Format for output file
- Example
chr pos tag1 tag2 tag3 Chr1 111403 0.05 nan 0.02 Chr1 111500 1.00 0.80 0.60 Chr2 20000 0.96 0.33 0.66- Column Description
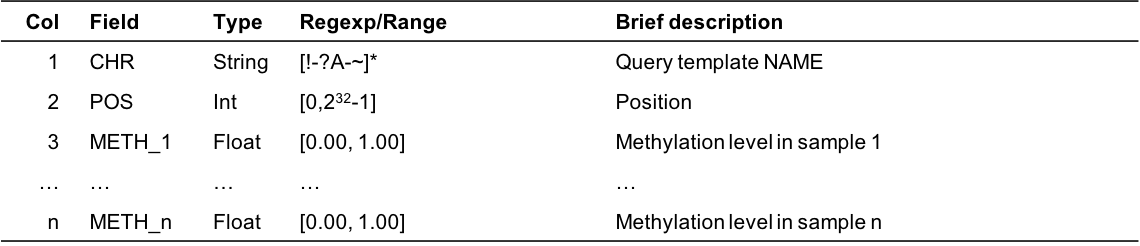
Figure 3.3: Output format description for cgmaptools fill tomatrix
3.6.2 mergelist tosingle
- Command
cgmaptools mergelist tosingle -h# Usage: cgmaptools mergelist tosingle -i f1,f2,..,fn [-o <output>]
# (aka MergeListOfCGmap)
# Description: Merge multiple CGmap/ATCGmap files into one.
# Contact: Guo, Weilong; guoweilong@126.com
# Last Update: 2018-04-10
# Note: Large memory is needed.
# Split input by chromosome for merge will save some memory.
#
#
# Options:
# -h, --help show this help message and exit
# -i FILE List of input files; gzipped file ends with '.gz'; seperated by
# comma without gap
# -f FILE cgmap or atcgmap [Default: cgmap]
# -o OUTFILE To standard output if not specified; gzipped file if end with
# '.gz'- Example
3.7 sort
- Command
cgmaptools sort -h# Usage: Sort_chr_pos [-i <input>] [-c 1] [-p 3] [-o output]
# Author : Guo, Weilong; guoweilong@gmail.com; 2014-05-11
# Last Update: 2018-01-02
# Description: Sort the input files by chromosome and position.
# The order of chromosomes would be :
# "chr1 chr2 ... chr11 chr11_random ... chr21 ... chrM chrX chrY"
#
# Options:
# -h, --help show this help message and exit
# -i FILE File name end with .CGmap or .CGmap.gz. If not specified,
# STDIN will be used.
# -c INT, --chr=INT The column of chromosome [default: 1]
# -p INT, --pos=INT The column of position [default: 2]
# -o OUTFILE To standard output if not specifiedExample
zcat RR*.CGmap.gz WG.CGmap.gz | gawk '$8>=5' | cut -f1,3 | sort -u | cgmaptools sort -c 1 -p 2 > index
3.8 split
- Command
cgmaptools split -h# Usage: cgmaptools split -i <input> -p <prefix[.chr.]> -s <[.chr.]suffix>
# (aka CGmapSplitByChr)
# Description: Split the files by each chromosomes.
# Contact: Guo, Weilong; guoweilong@126.com
# Last Update: 2018-01-02
#
# Options:
# -h, --help show this help message and exit
# -i FILE Input file, CGmap or ATCGmap foramt, use STDIN when not
# specified.(gzipped if end with 'gz').
# -p STRING The prefix for output file
# -s STRING The suffix for output file (gzipped if end with 'gz').Example
cgmaptools split -i WG.CGmap.gz -p WG -s CGmap.gz
3.9 select
- Command
cgmaptools select -h# Usage: cgmaptools select <command> [options]
# Version: 0.1.2
# Updated on: Dec. 14th, 2018
# Commands:
# region select or exclude liens by region lists
# site select or exclude lines by site list3.9.1 select region
- Command
cgmaptools select region -h# Usage: cgmaptools select region [-i <CGmap/ATCGmap>] -r <BED> [-R]
# (aka CGmapSelectByRegion)
# Description: Lines in input CGmap/ATCGmap be selected/excluded by BED file.
# Strand is NOT considered.
# Output to STDOUT in same format with input.
# Contact: Guo, Weilong; guoweilong@126.com
# Last Update: 2016-12-07
# Options:
# -i Input, CGmap/ATCGmap file; use STDIN if not specified
# Please use "gunzip -c <input>.gz " and pipe as input for gzipped file.
# Ex: chr12 G 19898796 ...
# -r Input, Region file, BED file to store regions
# At least 3 columns are required
# Ex: chr12 19898766 19898966 XX XXX XXX
# -R [optional] Reverse selection. Sites in region file will be excluded when specified
# -h help
# Tips: program will do binary search for each site in regionsExample
for CHR in 1 2 3 4 5; do (for P in 1 2 3 4 5; do echo | gawk -vC=$CHR -vP=$P -vOFS="\t" '{print "chr"C, P*1000, P*1000+200, "+";}' ; done) ; done > region.bedzcat WG.CGmap.gz | cgmaptools select region -r region.bed | head
3.9.2 select site
- Command
cgmaptools select site -h# Usage: cgmaptools select site -i <index> [-f <CGmap/ATCGmap>] [-r] [-o output]
# (aka CGmapSelectBySite)
# Description: Select lines from input CGmap/ATCGmap in index or reverse.
# Contact: Guo, Weilong; guoweilong@126.com
# Last Update: 2016-12-07
# Index format example:
# chr10 100504
# chr10 103664
#
# Options:
# -h, --help show this help message and exit
# -i FILE Name of Index file required (gzipped if end with '.gz').
# -r reverse selected, remove site in index if specified
# -f STRING Input CGmap/ATCGmap files. Use STDIN if not specified
# -o STRING CGmap, Output file name (gzipped if end with '.gz').Example
gawk 'NR%100==50' index > sitecgmaptools select site -f RR.CGmap.gz -i site -o RR_select.CGmap.gz