8 Other Ultilities
8.1 findCCGG
- Command
cgmaptools findCCGG -h# Usage: cgmaptools findCCGG -i <genome.fa> [-o <output>]
# (aka FiindCCGG)
# Description: Get the positions of all the C'CGG---CCG'G fragments.
# Contact: Guo, Weilong; guoweilong@126.com
# Last Update: 2018-01-02
# Output Ex:
# chr1 4025 5652
# chr1 8274 8431
#
# Options:
# -h, --help show this help message and exit
# -i FILE Genome sequence file in Fasta format
# -o FILE Name of the output file (standard output if not specified).
# Format: chr cCgg_pos ccGg_pos (0-base)Example
cgmaptools findCCGG -i genome.fa -o genome.ccgg
8.2 bed2fragreg
- Command
cgmaptools bed2fragreg -h# Usage: cgmaptools bed2fragreg [-i <BED>] [-n <N>] [-F <50,50,..> -T <50,..>] [-o output]
# (aka FragRegFromBED)
# Description: Generate fragmented regions from BED file.
# Contact: Guo, Weilong; guoweilong@126.com
# Last Update: 2018-05-02
# Split input region into N bins, get fragments from 5' end and 3' end.
# Input Ex:
# chr1 1000 2000 +
# chr2 9000 8000 -
# Output Ex:
# chr1 + 940 950 1000 1200 1400 1600 1800 1850
# chr2 - 9060 9050 9000 8800 8600 8400 8200 8150
#
#
# Options:
# -h, --help show this help message and exit
# -i FILE BED format, STDIN if omitted
# -F INT_list List of region lengths in upstream of 5' end, Ex: 10,50. List
# is from 5'end->3'end
# -T INT_list List of region lengths in downstream of 3' end, Ex: 40,20. List
# is from 5'end->3'end
# -n INT Number of bins to be equally split [Default:1]
# -o OUTFILE To standard output if omitted. Compressed output if end with
# .gzExample
Output format
- Example
chr1 + 940 950 1000 1200 1400 1600 1800 2000 2060 2080 chr2 - 9060 9050 9000 8800 8600 8400 8200 8000 7960 7940- Column Description
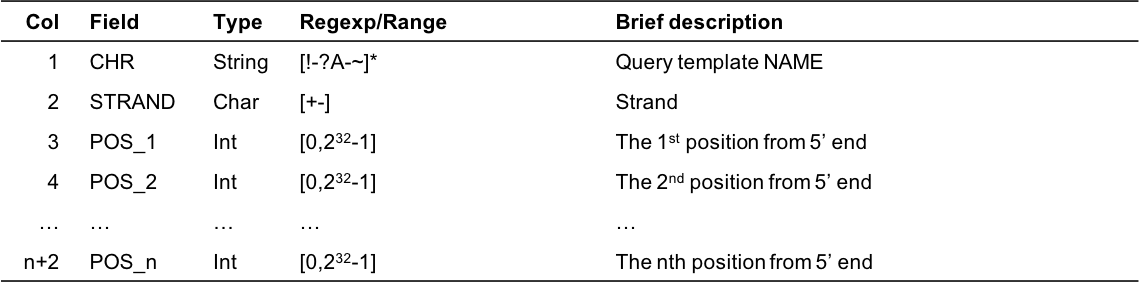
Figure 8.1: Output format description for cgmaptools bed2fragreg
[POS_1, POS_2), [POS_2, POS_3), … [POS_(n-1), POS_n) will be used as input for cgmaptools mfg